
Highly concentrated electrolytes offer enhanced energy-density for aqueous batteries, but the high salt concentration presents formidable challenges for practical implementation. Now, an electrolyte has been designed that has a substantially reduced salt concentration while still enabling high-performance batteries.
Driven by cost, as well as concerns over the toxicity and safety of organic lithium-ion batteries (LIBs), aqueous LIBs were proposed as early as 1994; however, they have never been commercialized owing to their limited energy density1. This is because the cell potential of aqueous LIBs is limited by the thermodynamic stability window of water (1.23 V), outside of which water is electrolysed. To expand the voltage window, water-in-salt electrolytes (WiSE)—solutions in which Li-salts (often sulfonimide-based) outweigh water in mass and volume—have been proposed as an effective strategy. In these systems, a solid electrolyte interphase (SEI) forms, passivating the negative electrode. The use of highly concentrated salts (>20 mol kg–1 or 20 m), however, prevents their practical application2. Now, writing in Nature Energy, Chunsheng Wang and co-workers from the University of Maryland demonstrate3 that practical LIB pouch cells can be assembled using aqueous electrolytes having a salt concentration below 5 m. Their approach not only alleviates the cost of using highly concentrated salts, but also demonstrates a new way to passivate the negative electrode, thus expanding the voltage window and improving the energy density.
Expanding the stability window of aqueous LIBs hinges on controlling the passivation mechanism at the negative electrode, which faces two main challenges. First, Li-salts are unable to decompose in water to form the counter anions necessary for the precipitation of Li-based inorganic compounds. Second, the solubility of such Li-based inorganic products is generally greater in aqueous than in organic electrolytes, further preventing the passivation.
The peculiar solvation structure found in WiSE triggers the reactivity of sulfonimide counterions to donate fluorine anions, eventually forming a LiF-rich SEI, whose solubility is low due to the common ion effect (Fig. 1). Although theoretically elegant, the use of WiSE does not completely suppress water reduction at negative electrodes, and consequently the batteries inherently suffer from fast self-discharge and low coulombic efficiencies (CE)4,5. Furthermore, the use of high concentrations of corrosive salts (>20 m) is in direct contradiction with the initial goal of reducing cost and improving safety in the development of aqueous LIB batteries.
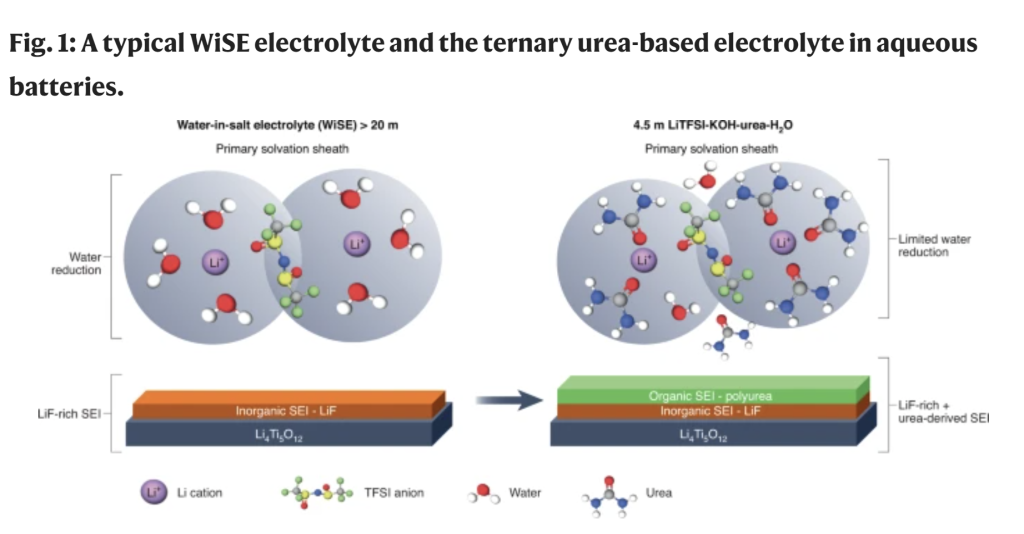
In their work, Wang and team assembled a 4.5 m LiTFSI-KOH-CO(NH2)2-H2O electrolyte (LiTFSI is lithium bis(trifluoromethanesulfonyl)imide) utilizing a combination of different strategies previously proposed. First, the researchers used CO(NH2)2, more commonly known as urea6, as a diluent to form strong interactions with Li+ and water. This diluent disrupts the hydrogen bonding network, which in diluted systems is responsible for the reactivity of water at electrochemical interfaces7. The use of urea therefore reduces the high concentration of the LiTFSI salt typically used in WiSE (that is, ~20 m).
Furthermore, a urea polymerization product forms on the surface of the negative electrode, providing organic functionality to the SEI that complements the LiF formed through the decomposition of TFSI anions (Fig. 1). To trigger this latter decomposition, which is normally only achieved in WiSE systems for aqueous LIBs, KOH was added to catalyse TFSI– degradation previously shown to be unstable in strongly alkaline conditions8. This mixed inorganic–organic SEI provides greater stability compared with the SEI formed in WiSE systems, as demonstrated by electrochemical measurements.
Wang and team assessed the performance of this electrolyte using LiMn2O4 and Li4Ti5O12 as positive and negative electrodes, respectively, both at the laboratory scale (that is, coin cells) as well as in a more practical format by assembling pouch cells. Using the aforementioned electrolytes, the researchers showed LiMn2O4||Li4Ti5O12 full cells with a CE of ~99.9%. This performance metric represents a neat gain compared with the 99% CEs previously obtained, for instance, for LiFePO4||Mo6S8 in 20 m LiTFSI (ref. 5) and LiMn2O4||Li4Ti5O12 using molecular crowding electrolytes9. This result is significant, because for coin cells an improvement of 99% to 99.9% CE theoretically increases the cycle life by one order of magnitude4.
Next, the Wang group assessed the performance in pouch cells with an areal capacity of 2.5 mAh cm–2, achieving a 99.87% CE and a 72% capacity retention over 500 cycles. They also examined the self-discharge rate and found a capacity decay of 18.7% per month. This is comparable to LiMn2O4||Li4Ti5O12 pouch cells with conventional organic electrolytes and commercially available Ni-MH batteries. Wang and team attribute this performance to the aforementioned stable SEI formation and inhibited water reduction.
Several roadblocks remain to be alleviated before envisaging this battery chemistry for practical usage. Despite reducing the amount of salts while improving the CE, using this complex electrolyte in fact lowers the amount of water in the system. Hence, given the values reported in this work (CE, electrode and electrolyte loadings), the concentration of water would be reduced after a limited number of cycles following the model proposed by Kühnel et al4. Such water loss could alter cell operation and its performance. To avoid this drying out of the cell, H2 gas generated during self-discharge and/or cycling must be controlled so that the water concentration can be maintained.
In conclusion, this work by Wang and co-workers reports an ingenious liquid electrolyte engineering approach that improves the cell performance of aqueous batteries. No doubt that future work will focus on tackling the remaining issues in order to assess the practicality of this novel chemistry.
Original Article: With only a grain of salt



